Importing GBS Files
TASSEL is a software widely used in the plant genetics community. This software inputs and outputs a variety of file formats. A common input and output format is the HapMap format, and there two versions, one in which genotypes are represented by two letters and it is called .hapdip.hmp and the other in which genotypes are represented by single coded letters. In both versions genotype designations follow IUPAC nomenclature Both formats can be imported into JMP Pro.
GBS text files are specialized tab-delimited text files used to store variant calls for markers across a genome for numerous individuals, or samples, from a study population. These files contain standardized headers and columns. An example GBS .hmp file, the ch01-g2f_2017_ZeaGBSv27_Imputed_AGPv4.hmp file, obtained from CyVerse Data Commons and downloaded from here, containing maize genotypes, and viewed within a text editor, is shown below:
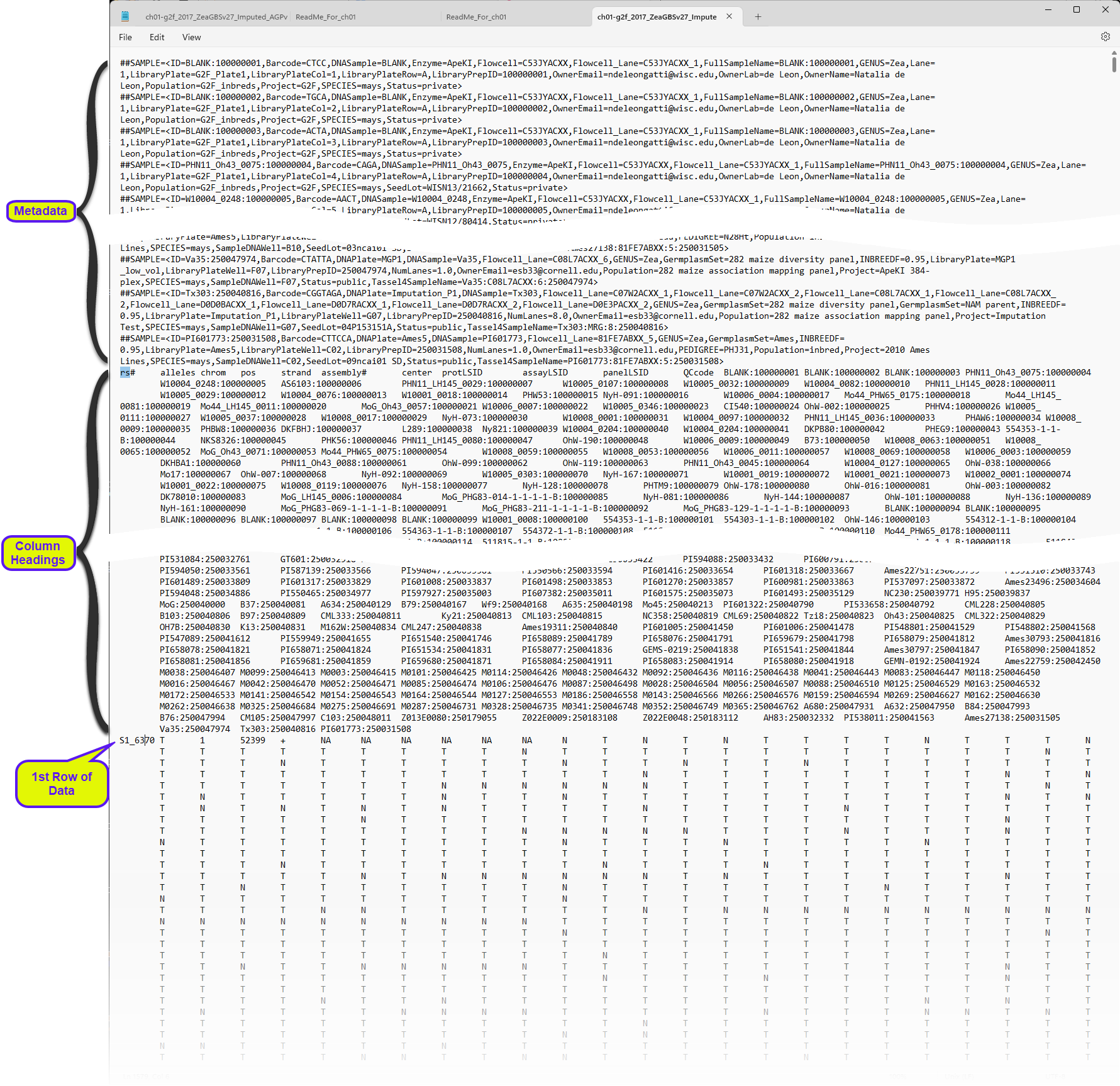
The file is structured very similar to VCF files, discussed previously. Rows at the beginning of the file contain metadata for the samples and genes contained in the file. The metadata is followed by a single row containing column headings, and then the genotype data.
In this format, single HapMap alleles are indicated by a single letter IUPAC nucleotide designation. The HapMap .hapdip.hmp file appears very similar. The only difference is that the genotypes of both alleles in the diploid pair are shown at each locus.

Importing HapMap (.hmp) Files
In this example, we import the ch01-g2f_2017_ZeaGBSv27_Imputed_AGPv4.hmp file shown above.
| 8 | Select File > Open. An Open Data File window opens to the last location used. |
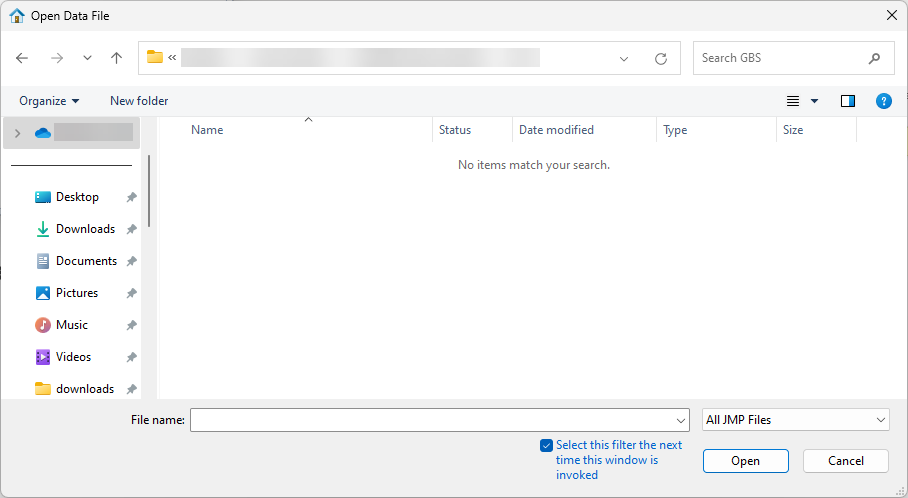
• Navigate to the location of the GBS file and use the drop-down menu (shown below) to select Genomics Text Files (*.txt; *.tsv; *.csv; *.bed; *.gff; *.gff3; *.gtf; *.mxt; *.vcf).
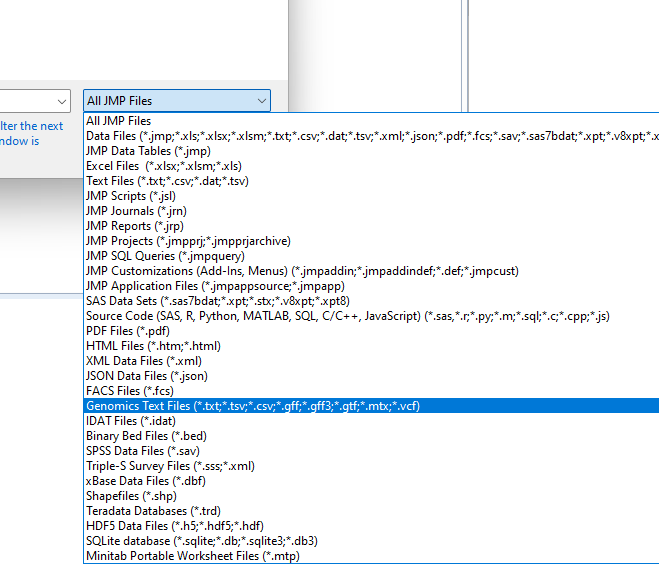
The options in the window change.
| 8 | Select the desired file. |
| 8 | Check the Open as Data (Using Preview) radio button. |
| 8 | Click . |
The Preview window is shown below:
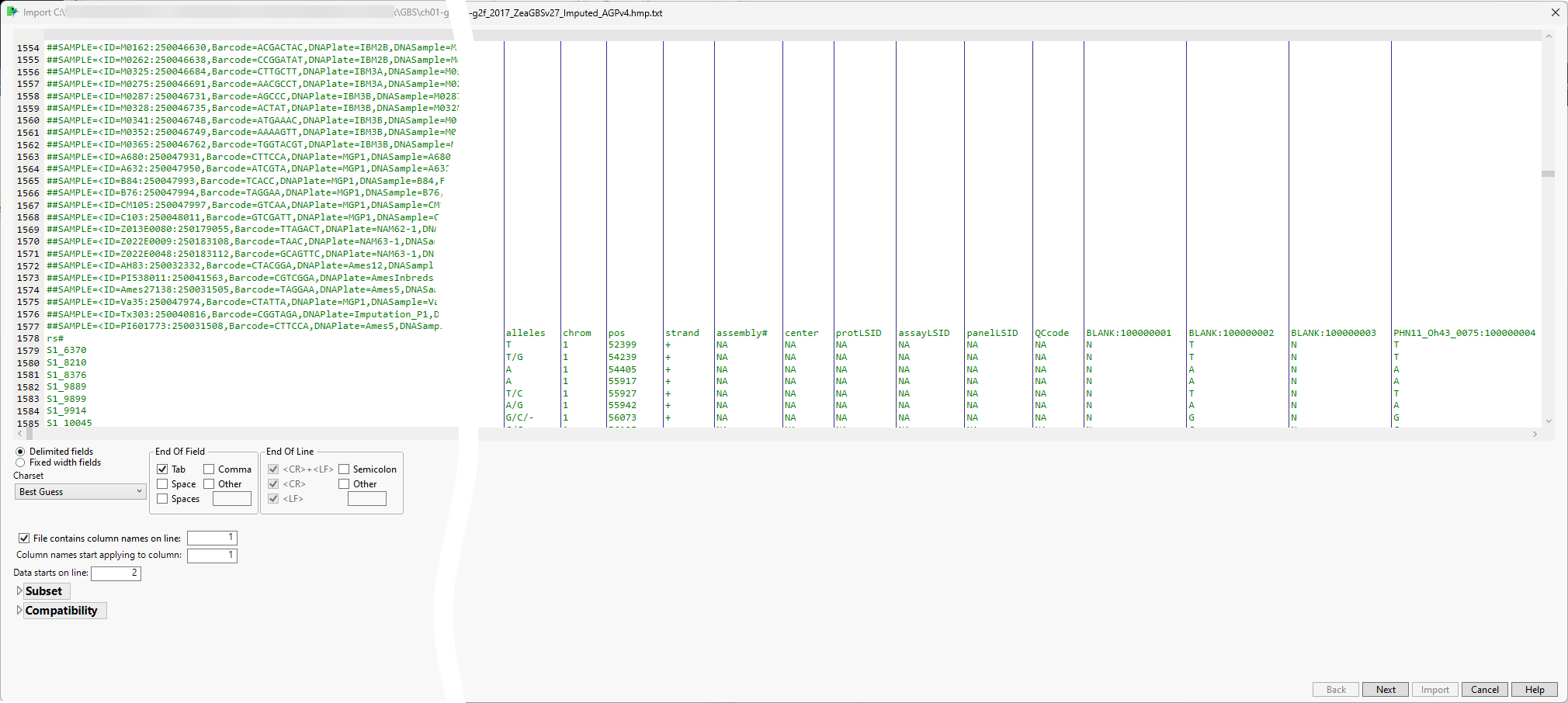
The preview enables you to visually inspect your data. You can identify the rows containing metadata and column names. Scroll through the table to determine the rows and columns containing the data. In this example, the column names are in row 1578 and the data starts in row 1579). Vertical bars in the table delimit the data included in each column.
The wizard also presents options for specifying features of and subsetting the data. Refer to Options in the JMP Text Import Window for more information about these options. In most cases, you will not need to change default settings for these options. However, you might need to specify the rows containing column names and the start of the data. (Note: The line the data starts on refers to the row imported.) Additionally, it might be useful to subset very large data sets into multiple JMP tables that can be joined later.
| 8 | Make sure the check box for the File column names on line: option is checked and enter the number of the row containing the column names (1578, in this example) in the text box, |
| 8 | Enter the number of the first row containing data (1579, in this example) in the Data starts on line text box. |
| 8 | Click . |
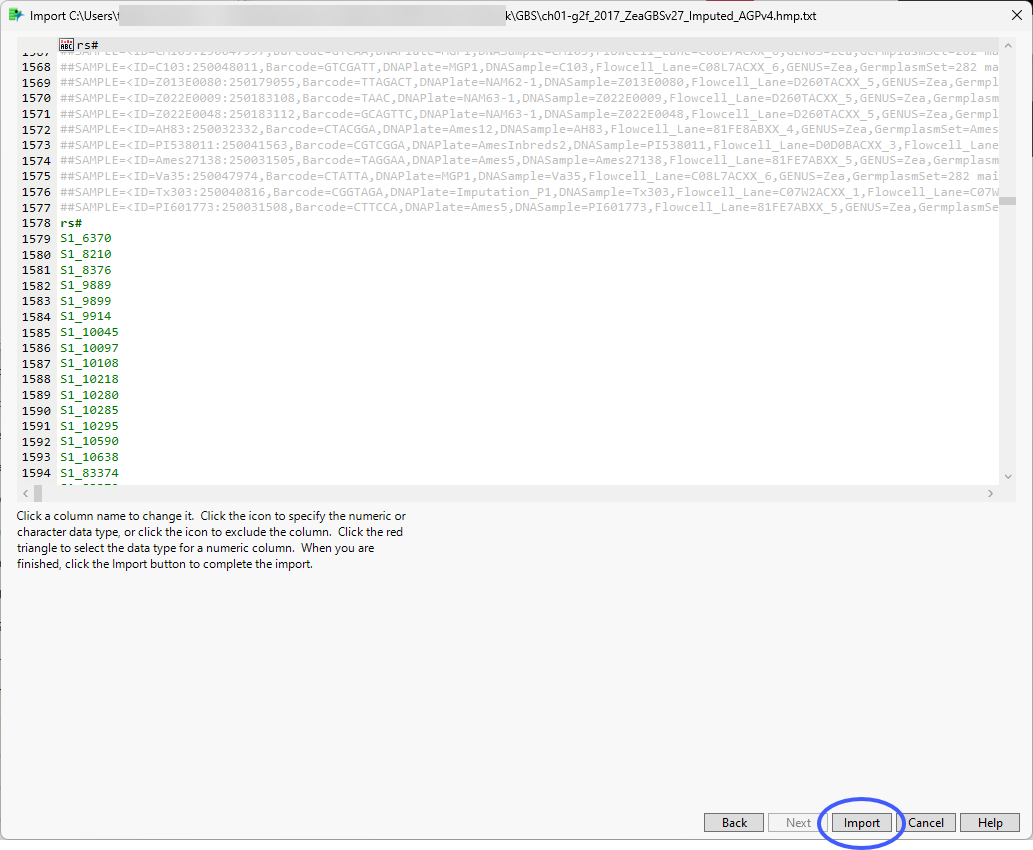
The metadata rows are grayed out and are not included in the JMP table.
| 8 | If everything is satisfactory, click to generate the JMP table. |
One table is generated by the import process: the ch01-2f_2017_ZeaGBSv27_Imputed_AGPv4.hmp.jmp table.
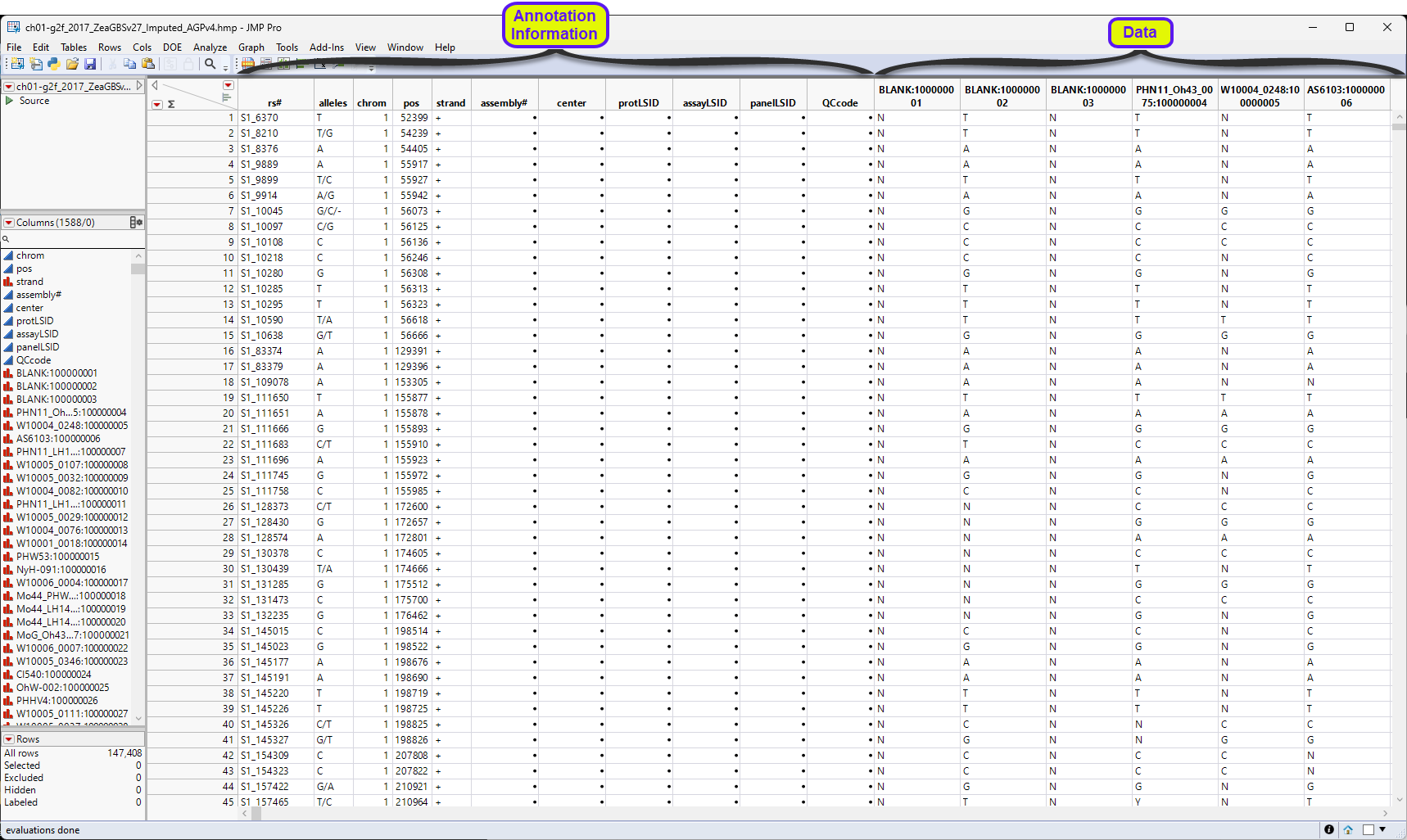
Before we continue, let us discuss how genomic data should be structured for analysis in JMP Pro. Most of the processes in JMP Pro assume that the input table has a particular data structure.
First, JMP PRO distinguishes between tall and wide data sets. A tall data table has samples as columns and molecular entity (for example, markers, genes, clones, proteins, or metabolites) as rows, whereas a wide data table is the transpose of the tall, having the samples as rows and molecular entities as columns.
When specifying the input data set for a process, it is important to know the required form. Most genomic analyses in JMP Pro require a wide data table. The Transpose platform under the Tables menu enables you to transform your data tables between tall and wide forms.
The ch01-2f_2017_ZeaGBSv27_Imputed_AGPv4.hmp.jmp table above is a tall data table. The first 11 columns contain annotation information (gene name, alleles, chromosome number and location, etc.). The data starts in column 12. Each column represents a sample. Individual genes are in rows. We cannot analyze the data in this format, so we must transpose it.
| 8 | Select Tables > Transpose to open the Transpose window. |
| 8 | Select all the data columns and drag them to the Transpose Columns box. |
| 8 | Select the rs# column (contains the marker names) as the Label column. |
| 8 | Note the default name of the transposed Output table name. Accept or change it as desired. |
| 8 | Specify a Label column name. |
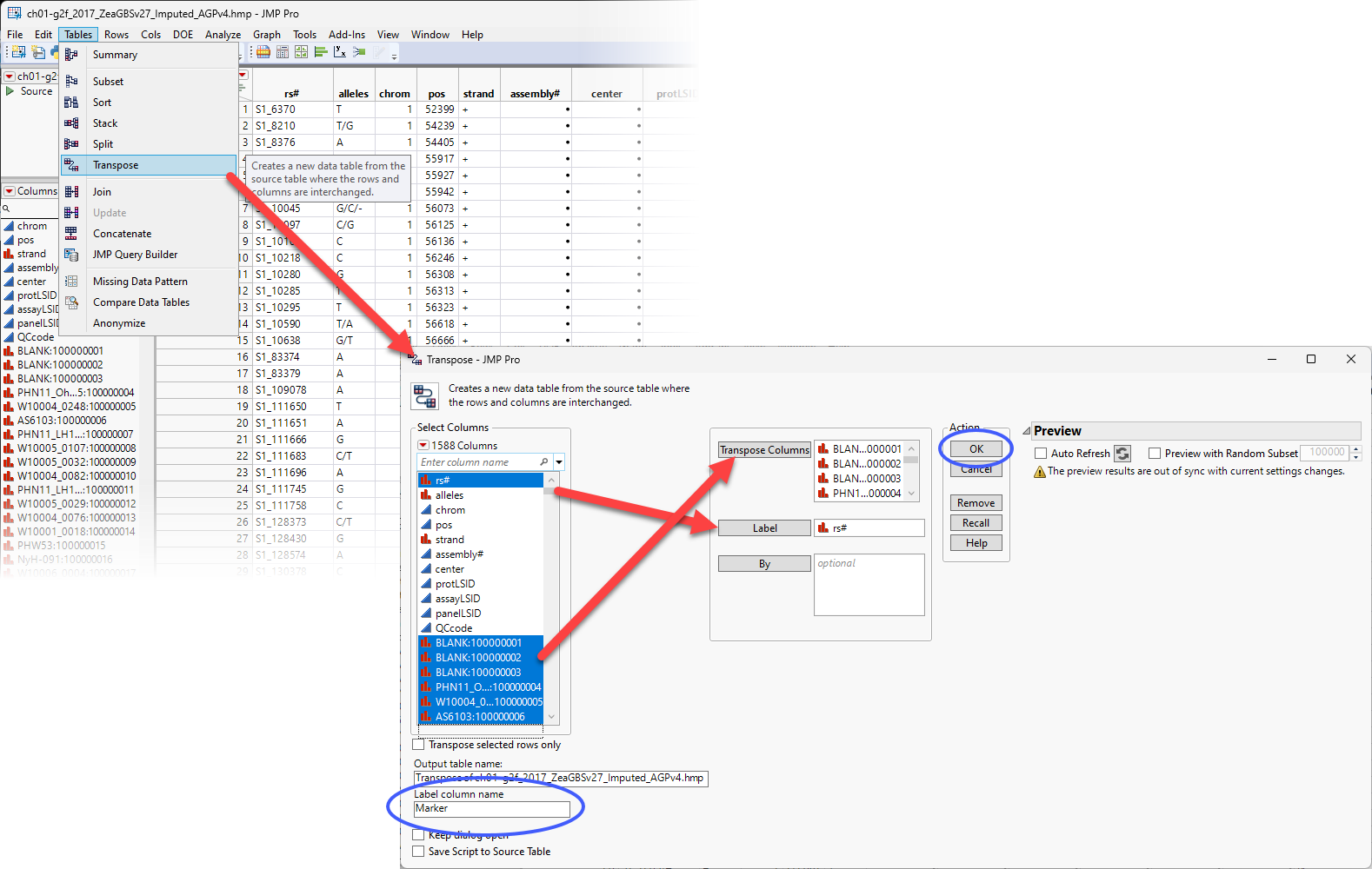
| 8 | Click . |
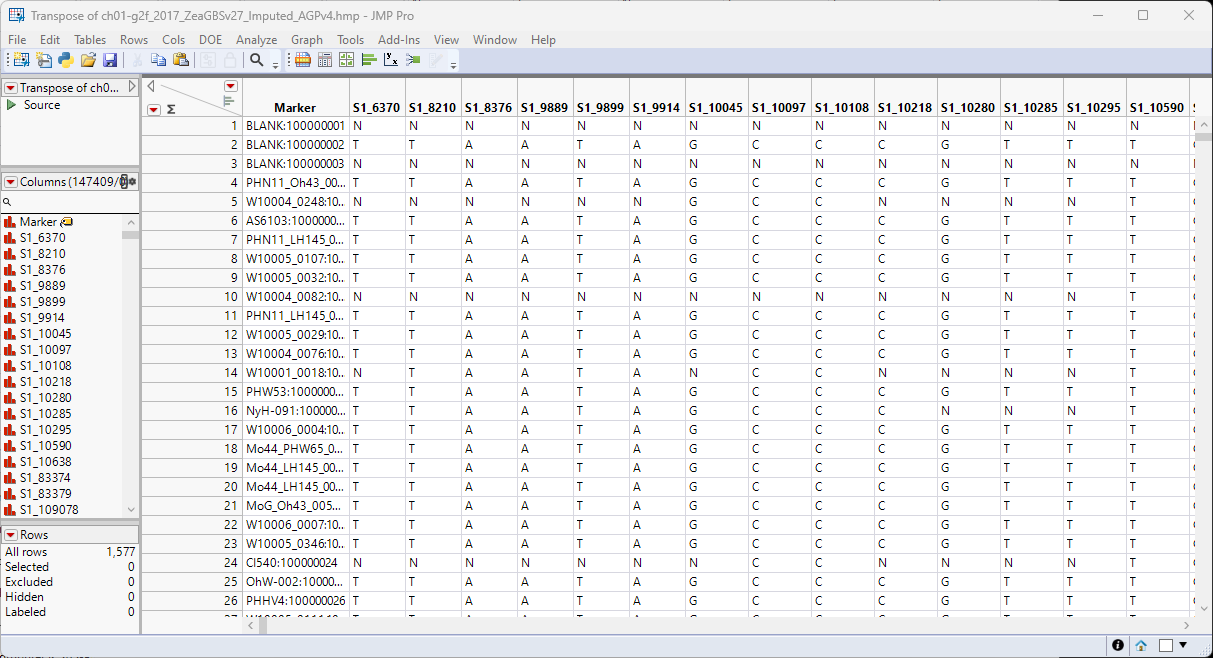
The Transpose of ch01-g2f_2017_ZeaGBSv27_Imputed_AGPv4.hmp.jmp transposed data table is a wide data table.
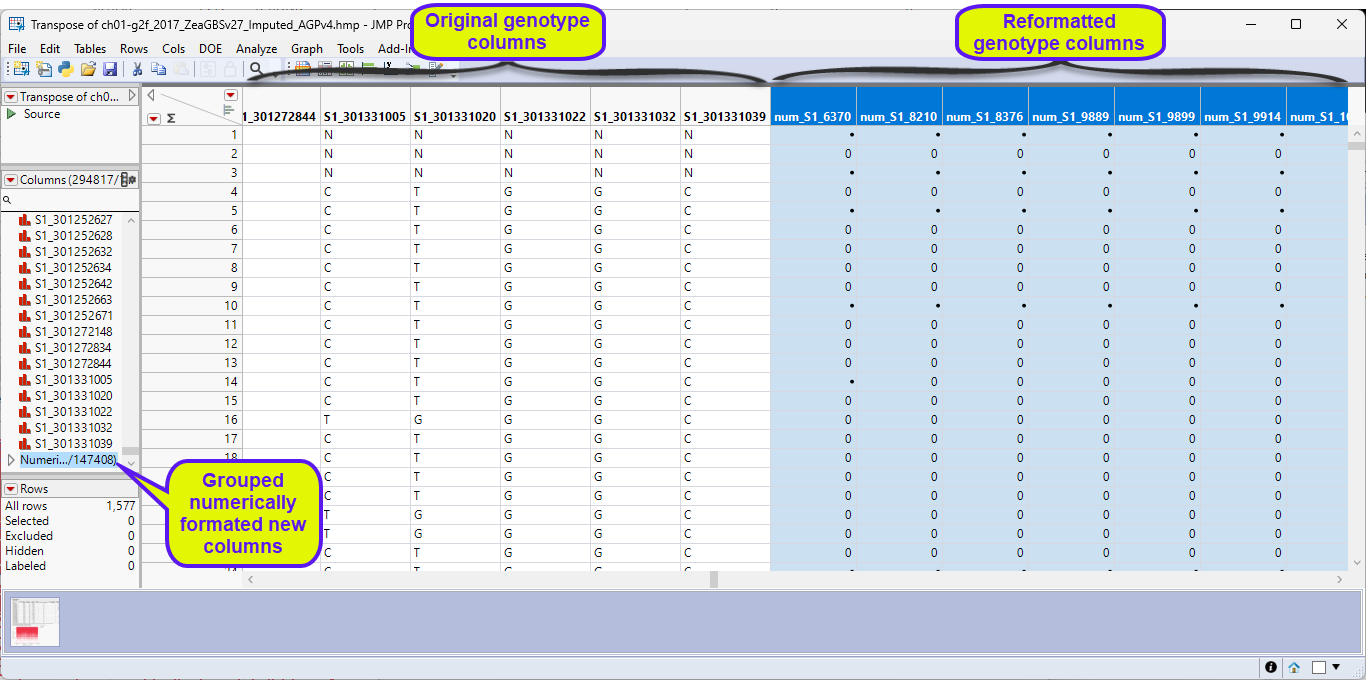
Another thing to consider is that marker data must be encoded in the one-column, numerical, genotypic format. Typically, in this format, diploid individuals homozygous for the least common, or minor allele, are represented in the table by a "2", whereas the heterozygotes are represented by a "1". Homozygotes for the most common allele are represented by a "0". This is not a common representation for genotypes. More typically, genotypes are represented by characters, either letters or numbers, often with both alleles represented with a delimiter. The genotype data in the Transpose of ch01-g2f_2017_ZeaGBSv27_Imputed_AGPv4.hmp.jmp table are represented by single-nucleotide characters. This format is not recognized by JMP Pro and must be recoded to the numerical form before we can proceed with the analysis. Fortunately, in JMP Pro v19, an option has been added to the Marker Statistics platform that reads and converts character and other formats and converts them to the one-column, numerical, genotypic format.
| 8 | Select Analyze > Genetics > Marker Statistics. |
| 8 | Select all the genotype columns as the Marker columns. |
| 8 | Use the drop-down menu to select Single Code Nucleotide as the Marker Format. |
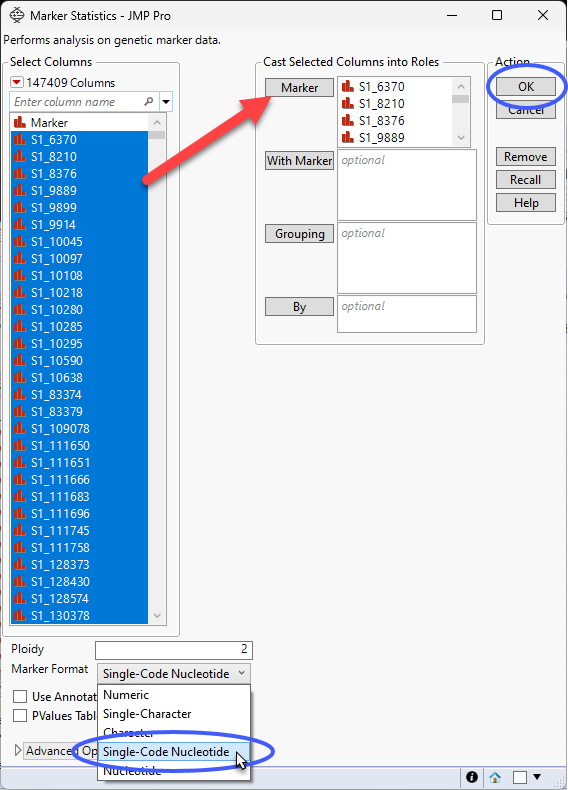
| 8 | Click . |
The Marker Statistics platform duplicates the columns, placing the new columns at the end of the table, and recodes the format of the genotypes to numeric and runs the Marker Statistics analysis (not shown here). The new columns are identified by the num_ prefix.
Importing HapMap .hapdip.hmp files
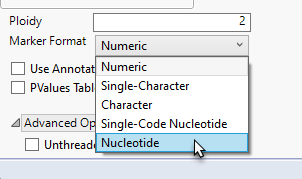
HapMap .hapdip.hmp files are imported exactly like HapMap .hmp files, as shown above. The only difference is during the recode step in Marker Statistics. Instead of selecting Single-Code Nucleotide as the Marker Format, you must choose Nucleotide.
Next Steps
For both .hmp and hapdip.hmp files, the resulting JMP data table is now ready for further analysis.
Note: In both cases, you must use the recoded numeric columns for any analysis.