Output Description
Basic Single Cell RNA-Seq Workflow
Running this process for the PBMC sample setting generates generates the tabbed Results window shown below. Refer to the Basic Single Cell RNA-Seq Workflow process description for more information. Output from the process is organized into tabs. Each tab contains one or more plots, data panels, data filters, and so on. that facilitate your analysis.

The Results window contains the following panes:
Tab Viewer
This pane provides you with a space to view individual tabs within the Results window. Use the tabs to access and view the output plots and associated data sets.
The following tabs are generated by this process:
| • | Data Overview: This tab displays several contour plots showing the total number of individual genes expressed in each cell, the total number of transcripts of all genes found in each cell, and the percentage of the counts in each cell made up by mitochondrial genes. Additional correlation plots show the correlations between total read counts and either the number of expressed genes or the percentage of expressed mitochondrial genes. |
| • | Variable Gene Selection: This tab displays a plot showing the average expression of genes across all cells verses the dispersion of genes that are being expressed. Selected variable genes are shown in red. |
| • | Clustering: This tab displays plots showing the clustering of individual cells into families based on the principle components analysis and either Hierarchical Clustering or K-Means Clustering. |
| • | Embedding: This tab diplays plots of the results of both tSNE and UMAP analysis of the cell clustering and gene expression. |
| • | Feature Screening: This tab displays a table and accompanying a bar chart representing the importance of the selected variable genes to the separation of cell groups. |
| • | Gene Expression Visualization: These tabs (one for marker genes and one for selected genes) each display three plots showing the log-transformed expression level of each gene across identified cell clusters by Hierarchical Clustering. A Column Switcher helps you to quickly navigate through the selected gene list. |
Follow-Up ANOVA
When the Launch ANOVA Interface check box is checked, the ANOVA process interface is launched as part of the output of the Basic Single Cell RNA-Seq Workflow process.
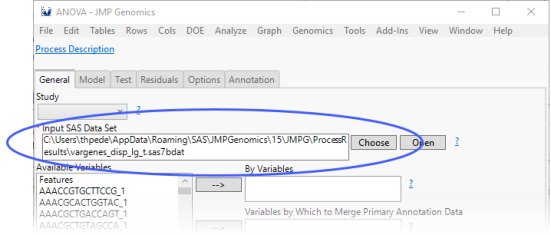
The output data (shown below) is preloaded as the input for the ANOVA.
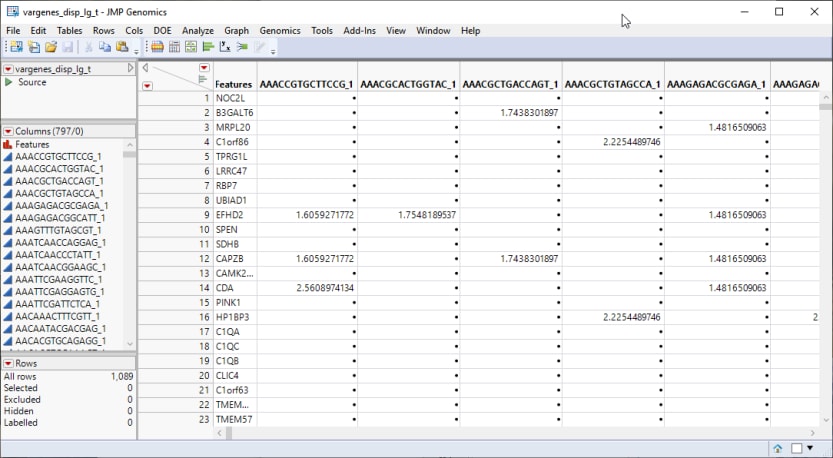
Note that the barcode gene identifiers now identify columns in this tall data set rather than rows in the original input data set.
This workflow added a column to the original PMBC.edf experimental design file identifying the cluster to which each gene belonged.
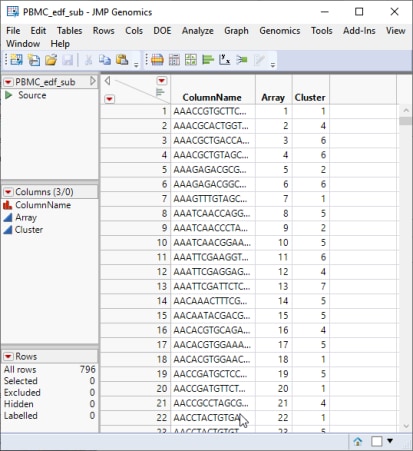
Note that the barcode gene identifiers identified in the ColumName column correspond to the columns in the ANOVA input data set.
This modified PBMC_edf_sub.sas7bdat experimental design file is specified in the ANOVA.

Running the the ANOVA process generates a pairwise comparison of all of the clusters. The volcano plots in the example shown here, show that expression of the S100A9 gene (circled in the plots in the figure below) is higher in cells in cluster 1 than the other clusters.
